
Happy Halloween! I was trying to think of something witty to connect DNA and Halloween, but have come up empty-handed. Or, perhaps a discussion of DNA sequencing and dredging up memories of past genetics coursework is frightening enough by itself for some of you out there. It still scares me sometimes too...
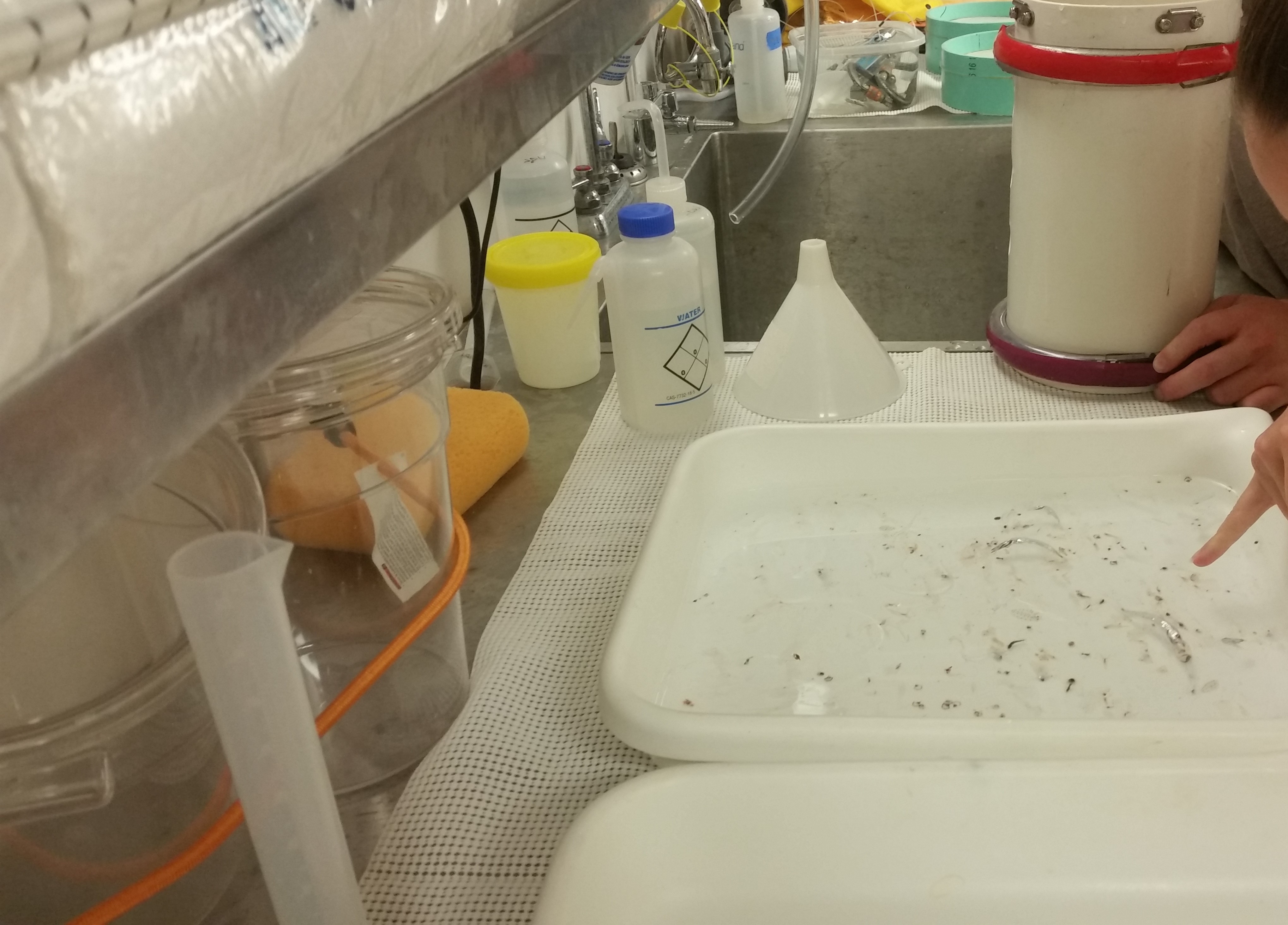
Once I take our filtered water samples back to the lab, there is a lot of work to do. We have to find a way to relate the biological diversity we saw, in this example zooplankton visually identified from a trawl sample (Figure 1), with sequence reads generated from a corresponding eDNA sample. How do we turn filtered water into actual sequences represented as strings of nucleotides (ATCGs)?
First, we extract the DNA from the water filters and quantify how much there is. Each filter takes a few hours to isolate the DNA from proteins and inhibitory compounds. Extreme care is taken not to cross-contaminate samples, or introduce ambient DNA from other projects (we don’t want butterfly or freshwater madtom DNA in our seawater samples). Next, we have a few options for sequencing, and the primary approach we are taking is called metabarcoding. If you’ve heard of the Barcode of Life Database (or BOLD), that basically represents an effort to sequence a species specific region of DNA for all of the world’s biodiversity, where each sequence serves as a “barcode” analogous to different barcodes on groceries at the store. If a researcher collects an organism in the field and sequences it at a gene in the BOLD database, they can compare it to the database and see if there is a match to their species of interest. If not, it may be a new species.
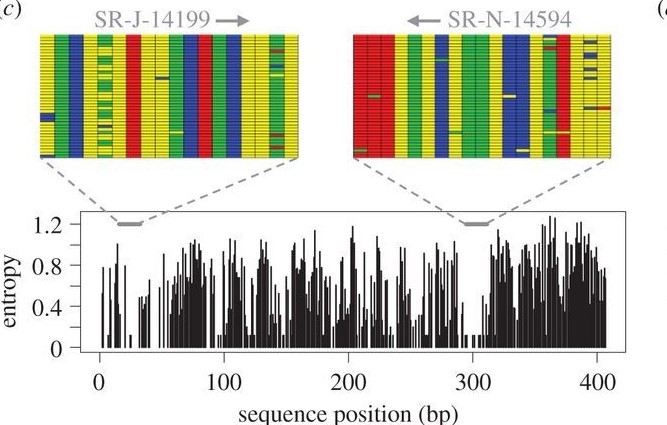
While the BOLD approach is species by species, metabarcoding seeks to sequence many different species at one time. Figure 2 attempts to illustrate primer design for this approach. Basically, we design a set of DNA primers that target a conserved portion of DNA flanking variable regions within. These primers are designed to broadly amplify things like fish or diatoms (not both), but usually incidentally pick up other things too like Bacteria. We take our extracted eDNA, and use polymerase chain reaction (PCR) to amplify anything that is “picked up” by these primers – hopefully mostly our target of interest. The resulting amplified DNA soup is put on a high throughput sequencer, which turns the DNA into sequence reads in a file that can be analyzed on a computer. We then compare the millions of sequence reads against a database of things we expect to see, and assign a taxonomy when appropriate. The end result is numbers of reads assigned to different taxa - a community profile.
One important part of this endeavor is building up our reference database, or grocery store inventory – after all, if a creature is not represented in the database, it can be difficult or impossible to figure out what it is. Therefore, we are also taking back samples of different organisms collected from the net samples to enhance our reference database. Hopefully, at the end of all this, we will be getting close to the ability to take a seawater sample, sequence it, and see what is there – all from a liter of water.
Author: Aaron Aunins.
